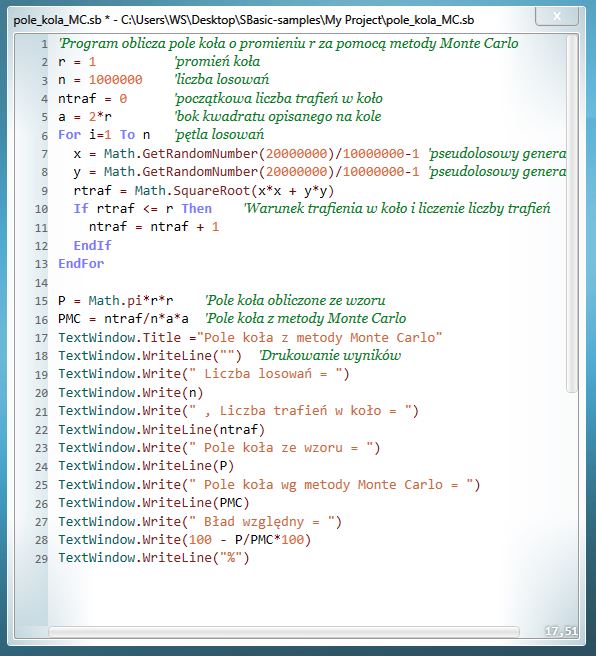
Poprzednia wprawka pozwoliła mi zrozumieć istotę metody Monte Carlo. Teraz postanowiłem zastosować ją do szkolnego, ale nieco bardziej zaawansowanego zagadnienia. Stany energetyczne atomu wodoru można obliczyć z wykorzystaniem wzorów Bohra lub równania Schroedingera. Tu zastosowałem metoda Monte Carlo, która jest popularnym w fizyce i chemii sposobem obliczania stanów rozmaitych układów kwantowych. W celu obliczenia stanu podstawowego atomu wodoru wykorzystałem algorytm opublikowany przez prof. A. Dawida (obecnie Wyższa Szkoła Biznesu w Dąbrowie Górniczej). Sporo czasu zajęło mi rozpracowanie oryginalnego algorytmu i zrozumienie działania jego składowych. Po kosmetycznych zmianach kodu zapisałem go w języku VBA dla Excela. Poniżej zapisany program oblicza zależność energii stanu podstawowego atomu wodoru od wartości zmiennej wariacyjnej. Minimalna wartość energii dla obliczenia ścisłego wynosi -0.5 hartree.


Dla funkcji psi postaci

Wynik działania hamiltonianu na tę funkcję wyraża się wzorem:

Wobec powyższego wzór na energię lokalną, używaną w algorytmie Monte Carlo, ma postać:

Fragment programu umieszczony w poprzednim wpisie, a odnoszący się do modułu całkowania (właściwie sumowania) upraszcza się do postaci:
W poprzednim poście pokazałem zapis modułu całkowania energii lokalnych z wykorzystaniem analitycznej postaci wyniku działania hamiltonianu dla atomu wodoru na funkcję exp(-cr). W niniejszym poście wykorzystałem to samo podejście i wyznaczyłem postać wyniku działania hamiltonianu dla atomu wodoru na funkcję gaussowską exp(-cr2):

Wzór na energię lokalną ma postać, jak niżej:

Funkcja programu QBASIC definiująca funkcję Gaussa w programie ma postać:
W celu dokładnego zrozumienia sposobu obliczania energii atomu wodoru z wykorzystaniem metody Monte Carlo i algorytmu Metropolisa, napisałem program w języku QBASIC. Program wykorzystuje znany generator liczb pseudolosowych o rozkładzie normalnym. Aby przekonać się, że tak się dzieje w rzeczywistości wyniki losowań zapisałem w pliku, z którego wykonałem wykres za pomocą programu Excel. W programie umieściłem kod, który pozwala zliczać losowania w wektorze x:


Uwaga. Ta aproksymacja nie jest zbyt dobra. Lepsza została pokazana w następnym wpisie.
Program generuje zbiór danych, które można przedstawić graficznie. Poniżej zamieściłem wykres uzyskany za pomocą programu Excel:

Jak widać na wykresie, algorytm Metropolisa traktuje promienie w sposób wybiórczy. Uzyskana krzywa przypomina funkcję dystrybucji radialnej (radialnej gęstości elektronowej) stanu podstawowego atomu wodoru z maksimum poniżej jedności (w obliczeniach użyłem jednostki atomowe).
Public c, pi As Double
Sub atomH() 'procedura oblicza energię stanu podstawowego atomu wodoru
Dim x, y, z, h, h2 As Double
pi = 3.14159265
llos = 1000000 'liczba losowań
h = 0.01 'krok różniczkowania
h2 = h * h 'kwadrat kroku różniczkowania
j = 1 'licznik rzędu w tablicy Excela
For c = 0.2 To 1.9 Step 0.1 'obl. energii w zal. od zm. wariacyjnej
En = 0 'zerowanie energii do sumowania
x = 0: y = 1.2: z = 0 'punkt początkowy
For i = 1 To llos 'pętla obliczeniowa
xzapas = x 'zabezpieczenie starych współrzędnych
yzapas = y
zzapas = z
psi = fufal(x, y, z) 'wartość psi w punkcie (x,y,z)
psikw = psi * psi 'kwadrat psi
'Losowanie nowego punktu (xn,yn,zn)
xn = 2 * Rnd - 1 'zakres (-1, 1)
yn = 2 * Rnd - 1
zn = 2 * Rnd - 1
pr = Sqr(xn * xn + yn * yn + zn * zn) 'promień punktu (xn,yn,zn)
skok = 0.6 * GRNF 'skok przypadkowy wg rozkładu Gaussa
x = x + xn * skok / pr 'poprawione współrzędne nowego punktu
y = y + yn * skok / pr
z = z + zn * skok / pr
psinowa = fufal(x, y, z) 'wartość psi w nowym punkcie
psinowakw = psinowa * psinowa
'obliczanie warunku przejścia łańcucha do nowego punktu
rel = psinowakw / psikw 'stosunek nowej i starej psi
If rel < 1 Then 'przejście bezwarunkowe gdy rel >= 1
If Rnd > rel Then 'przejście odrzucone
x = xzapas
y = yzapas
z = zzapas
psinowa = fufal(x, y, z)
End If
End If
'obliczanie sumy energii lokalnych
'obliczanie drugich pochodnych cząstkowych w punkcie (x,y,z)
fufal2 = 2 * fufal(x, y, z)
d1 = (fufal(x - h, y, z) - fufal2 + fufal(x + h, y, z)) / h2
d2 = (fufal(x, y - h, z) - fufal2 + fufal(x, y + h, z)) / h2
d3 = (fufal(x, y, z - h) - fufal2 + fufal(x, y, z + h)) / h2
Lappsi = -(d1 + d2 + d3) / 2 'druga poch. psi w punkcie (x,y,z)
V = -1 / (Sqr(x * x + y * y + z * z)) 'potencjal, tu psi/psi=1
En = En + Lappsi / psinowa + V 'sumowanie energii lokalnych
Next i
Cells(j, 1) = c
Cells(j, 2) = En / llos 'zapis energii średniej
j = j + 1
Next c
End Sub
Function fufal(x, y, z) 'oblicza wartość funkcji psi w punkcie x,y,z
r = Sqr(x * x + y * y + z * z)
fufal = Exp(-c * r)
End Function
Function GRNF() 'generuje liczbę pseudolosową z rozkładu Gaussa
r1 = Sqr(-Log(1 - Rnd))
GRNF = r1 * Sin(2 * pi * Rnd)
End Function
Wykres przedstawia wyniki obliczeń dla miliona losowań. Minimum znajduje się przy c = 1 i wynosi prawie dokładnie -0.5 hartree.
W poprzednim wpisie przedstawiłem algorytm obliczania energii atomu wodoru z wykorzystaniem metody Monte Carlo. Energie lokalne liczone były za pomocą numerycznego różniczkowania funkcji falowej w punkcie (x,y,z). Celem tego różniczkowania było obliczenie wartości laplasjanu w tym punkcie.
Ponieważ różniczkowanie takie można wykonać symbolicznie, procedurę można uprościć wpisując do programu symboliczną postać wyniku działania hamiltonianu na wybraną funkcję psi. Takie podejście jest mniej ogólne od numerycznego wyznaczania wartości laplasjanu, ale może ułatwić zrozumienie sposobu działania algorytmu Metropolisa.
Hamiltonian działający na funkcję psi ma postać (jednostki atomowe):
Ponieważ różniczkowanie takie można wykonać symbolicznie, procedurę można uprościć wpisując do programu symboliczną postać wyniku działania hamiltonianu na wybraną funkcję psi. Takie podejście jest mniej ogólne od numerycznego wyznaczania wartości laplasjanu, ale może ułatwić zrozumienie sposobu działania algorytmu Metropolisa.
Hamiltonian działający na funkcję psi ma postać (jednostki atomowe):
Dla funkcji psi postaci
Wynik działania hamiltonianu na tę funkcję wyraża się wzorem:

Wobec powyższego wzór na energię lokalną, używaną w algorytmie Monte Carlo, ma postać:
Fragment programu umieszczony w poprzednim wpisie, a odnoszący się do modułu całkowania (właściwie sumowania) upraszcza się do postaci:
...
'obliczanie cakłi z energii lokalnych
r1 = SQR(x * x + y * y + z * z)
En = En + (-c * c / 2 + (c - 1) / r1)
...
Wartość energii -0.5, jako minimum dla wartości c=1 otrzymuje się po dokonaniu sumowania 20000 konfiguracji.
Wzór na energię lokalną ma postać, jak niżej:
Funkcja programu QBASIC definiująca funkcję Gaussa w programie ma postać:
FUNCTION fufal (x, y, z)
r = SQR(x * x + y * y + z * z)
fufal = EXP(-c * r * r)
END FUNCTION
Odpowiedni moduł całkowania numerycznego sprowadza się do następujących instrukcji:...
'obliczanie calki z energii lokalnych
r1 = SQR(x * x + y * y + z * z)
El = -2 * c * c * r1 * r1 - 1 / r1 + 3 * c
En = En + El
...
Dla 500000 konfiguracji minimum dla c=0.3 wynosi -0.421 j.a., co w przeliczeniu na elektronowolty daje wartość -11.45 eV. Błąd wyliczenia w stosunku do dokładnej wartości energii wawynosi +15.8%.
W celu dokładnego zrozumienia sposobu obliczania energii atomu wodoru z wykorzystaniem metody Monte Carlo i algorytmu Metropolisa, napisałem program w języku QBASIC. Program wykorzystuje znany generator liczb pseudolosowych o rozkładzie normalnym. Aby przekonać się, że tak się dzieje w rzeczywistości wyniki losowań zapisałem w pliku, z którego wykonałem wykres za pomocą programu Excel. W programie umieściłem kod, który pozwala zliczać losowania w wektorze x:
'program GRNF generator lipselo o rozkladzie Gaussa
'zakres GRNF od -3.4 do 3.11 dla miliona losowan
pi = 3.14159265#
DIM x(-350 TO 350)
FOR i = 1 TO 100000
r1 = SQR(-LOG(1 - RND)) 'generowanie lipselo
GRNF = r1 * SIN(2 * pi * RND)
liczba = INT(GRNF * 100) 'zliczanie
x(liczba) = x(liczba) + 1
NEXT i
OPEN "gauss.dat" FOR APPEND AS #1
FOR i = -350 TO 350
PRINT #1, i, x(i) 'wpis wyniku do pliku
NEXT i
CLOSE
Zamieszczony poniżej wykres pokazuje, że generowane liczby pseudolosowe mieszczą się w zakresie od około -3.4 do około 3.2. Wartość średnia losowań jest bliska zera a maksymalna liczebność losowań średniej wynosi około 0.6% całkowitej liczby losowań:
Przedstawiony poprzednio wykres zależności częstości występowania promienia od jego wartości przeanalizowałem z wykorzystaniem postaci analitycznych funkcji. Kilka prób wykonanych za pomocą programu Derive pokazało, że dobrym przybliżeniem jest iloczyn funkcji Gaussa i kwadratu zmiennej niezależnej z odpowiednimi współczynnikami. Uzyskany wynik pokazuje poniższy wykres:

Uwaga. Ta aproksymacja nie jest zbyt dobra. Lepsza została pokazana w następnym wpisie.
Metoda Monte Carlo obliczania energii stanów atomu wodoru wykorzystuje algorytm Metropolisa wyboru punktów w przestrzeni wokółjądrowej. Dzięki temu wyborowi suma energii lokalnych aproksymuje energię całkowitą badanego stanu. Intrygowało mnie, które punkty są wybierane przez algorytm Metropolisa. Ze względu na fakt, że rozważany atom wodoru w stanie 1s jest kulistosymetryczny, wystarczyło zapytać, które wartości promienia wodzącego są wybierane i z jaką częstością.
W tym celu zmodyfikowałem program do obliczenia energii, umieszczony we Wprawce II. Usunąłem fragmenty obliczające energię lokalną i pozostawiłem tylko algorytm wyboru promienia. Dodałem procedurę zliczającą promienie. Zakres wektora liczności (6.2*100) ustaliłem eksperymentalnie dla 500000 losowań. Kod tak zmodyfikowanego programu ma następującą postać:
W tym celu zmodyfikowałem program do obliczenia energii, umieszczony we Wprawce II. Usunąłem fragmenty obliczające energię lokalną i pozostawiłem tylko algorytm wyboru promienia. Dodałem procedurę zliczającą promienie. Zakres wektora liczności (6.2*100) ustaliłem eksperymentalnie dla 500000 losowań. Kod tak zmodyfikowanego programu ma następującą postać:
DECLARE FUNCTION fufal! (x!, y!, z!)
DECLARE FUNCTION GRNF! () 'gaussowski generator pseudolosowy
'jednostki atomowe
CLS
SCREEN 12
llos = 500000 'liczba losowan
DIM SHARED c 'zmienna wariacyjna globalna
DIM x(620) 'wektor licznosci promieni
c = 1.5 'wybrana wartosc zm. wariacyjnej
OPEN "dane.DAT" FOR APPEND AS #1
x = 0: y = 1.2: z = 0 'poczatkowy punkt
FOR i = 1 TO llos
xzapas = x 'zabezpieczenie starych wspolrzednych
yzapas = y
zzapas = z
psi = fufal(x, y, z) 'wartosc psi w punkcie (x,y,z)
psikw = psi * psi 'kwadrat psi
'Losowanie nowego punktu (xn,yn,zn)
xn = 2 * RND - 1 'zakres (-1, 1)
yn = 2 * RND - 1
zn = 2 * RND - 1
pr = SQR(xn * xn + yn * yn + zn * zn) 'promien punktu (xn,yn,zn)
skok = .6 * GRNF 'skok przypadkowy wg rozkladu Gaussa
x = x + xn * skok / pr 'poprawione wspolrzedne nowego punktu
y = y + yn * skok / pr
z = z + zn * skok / pr
psinowa = fufal(x, y, z) 'wartsc psi w nowym punkcie
psinowakw = psinowa * psinowa
'obliczanie warunku przejscia lancucha do nowego punktu
rel = psinowakw / psikw 'stosunek nowej i starej psi
IF rel < 1 THEN 'przejscie bezwarunkowe gdy rel >= 1
IF RND > rel THEN 'przejscie odrzucone
x = xzapas 'przywrocenie starych wartosci
y = yzapas
z = zzapas
psinowa = fufal(x, y, z)
END IF
END IF
rn = SQR(x * x + y * y + z * z) 'promien do zliczania
liczba = INT(rn * 100) ' definicja adresu wektora promieni
x(liczba) = x(liczba) + 1 ' zliczanie promieni
NEXT i
FOR i = 1 TO 620
PRINT #1, i, x(i)
NEXT i
CLOSE
END
FUNCTION fufal (x, y, z)
r = SQR(x * x + y * y + z * z)
fufal = EXP(-c * r)
END FUNCTION
FUNCTION GRNF
r1 = SQR(-LOG(1 - RND))
GRNF = r1 * SIN(2 * 3.14159265# * RND)
END FUNCTION
Program generuje zbiór danych, które można przedstawić graficznie. Poniżej zamieściłem wykres uzyskany za pomocą programu Excel:

Jak widać na wykresie, algorytm Metropolisa traktuje promienie w sposób wybiórczy. Uzyskana krzywa przypomina funkcję dystrybucji radialnej (radialnej gęstości elektronowej) stanu podstawowego atomu wodoru z maksimum poniżej jedności (w obliczeniach użyłem jednostki atomowe).