Wzór wyrażający wartość średnią energii dla znormalizowanych i rzeczywistych funkcji falowych ma postać 1.
 Dla układów o symetrii kulistej element dv może być wyrażony, jako różniczka 2 wzoru określającego objętość kuli
Dla układów o symetrii kulistej element dv może być wyrażony, jako różniczka 2 wzoru określającego objętość kuli

 Po wstawieniu 2 do 1, pomnożeniu prawej strony przez stosunek
Po wstawieniu 2 do 1, pomnożeniu prawej strony przez stosunek
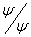 i uporządkowaniu wyrazów otrzymujemy wzór 3 na całkowitą, średnią energię układu. Wzór zawiera tzw. „energię lokalną”
i uporządkowaniu wyrazów otrzymujemy wzór 3 na całkowitą, średnią energię układu. Wzór zawiera tzw. „energię lokalną”
 oraz fragment będący wyrażeniem na funkcję gęstości radialnej.
oraz fragment będący wyrażeniem na funkcję gęstości radialnej.
 Po przejściu od całkowania do sumowania otrzymujemy wzór 4 na przybliżoną wartość energii układu wyrażoną przez (radialną) gęstość elektronową, energię lokalną i liczbę punktów n w przestrzeni wokółjądrowej, w których dokonujemy obliczeń a następnie sumowania.
Po przejściu od całkowania do sumowania otrzymujemy wzór 4 na przybliżoną wartość energii układu wyrażoną przez (radialną) gęstość elektronową, energię lokalną i liczbę punktów n w przestrzeni wokółjądrowej, w których dokonujemy obliczeń a następnie sumowania.
 Z drugiej strony do obliczenia energii z wykorzystaniem metody Monte Carlo i algorytmu Metropolisa stosuje się wzór 5.
Z drugiej strony do obliczenia energii z wykorzystaniem metody Monte Carlo i algorytmu Metropolisa stosuje się wzór 5.
 Sugerujemy, że wzory 4 i 5 są równoważne w tym sensie, że algorytm Metropolisa z wykorzystaniem wartości stosunku „nowej” i „starej” wartości funkcji gęstości elektronowej
Sugerujemy, że wzory 4 i 5 są równoważne w tym sensie, że algorytm Metropolisa z wykorzystaniem wartości stosunku „nowej” i „starej” wartości funkcji gęstości elektronowej
 jako kryterium wyboru przejścia do nowego punktu, faworyzuje pewne wartości promienia r, w których oblicza się energie lokalne. Innymi słowy, pewna wartość promienia może być wykorzystywana wielokrotnie do obliczenia gęstości a następnie wartości energii lokalnej. Zjawiska tego nie widać explicite we wzorze 5. W celu rozpoznania tego zagadnienia, rozpatrzmy model atom wodoru w przybliżeniu Borna-Oppenheimera i nieskończenie ciężkiego jądra. Stacjonarne równanie Schroedingera przyjmuje w jednostkach atomowych postać 6:
jako kryterium wyboru przejścia do nowego punktu, faworyzuje pewne wartości promienia r, w których oblicza się energie lokalne. Innymi słowy, pewna wartość promienia może być wykorzystywana wielokrotnie do obliczenia gęstości a następnie wartości energii lokalnej. Zjawiska tego nie widać explicite we wzorze 5. W celu rozpoznania tego zagadnienia, rozpatrzmy model atom wodoru w przybliżeniu Borna-Oppenheimera i nieskończenie ciężkiego jądra. Stacjonarne równanie Schroedingera przyjmuje w jednostkach atomowych postać 6:
 Wybieramy funkcję próbną postaci
Wybieramy funkcję próbną postaci
 Dla wybranego modelu metoda Monte Carlo i algorytm Metropolisa generują zbiór wartości promieni r (program liczno.bas) o charakterystyce przedstawionej na Rys. 1. dla 500000 losowań i dla c = 1.0.
Dla wybranego modelu metoda Monte Carlo i algorytm Metropolisa generują zbiór wartości promieni r (program liczno.bas) o charakterystyce przedstawionej na Rys. 1. dla 500000 losowań i dla c = 1.0.
Graficzna prezentacja wyniku działania programu

Wykres ten ilustruje anonsowany wyżej fakt, że energie lokalne obliczone dla danej wartości r są sumowane tyle razy, ile razy dany promień został wybrany podczas błądzenia. Aproksymacja przedstawionego zbioru punktów za pomocą funkcji opartej na funkcji gęstości radialnej daje wzór postaci 7.
 Otrzymana zależność 7 zgadza się, co do postaci, ze wzorem określającym funkcję gęstości radialnej z wyjątkiem wartości przedwykładniczego współczynnika liczbowego. Liczba 20000 znacznie przekracza oczekiwaną teoretycznie wielkość 4*pi. Ten wynik można zinterpretować, jako koszt obliczeniowy poniesiony w związku z zastosowaniem metody Monte Carlo i błądzenia wedle algorytmu Metropolisa, gdyż dokładność obliczenia energii całkowitej układu wzrasta wraz ze wzrostem liczby kroków błądzenia.
Otrzymana zależność 7 zgadza się, co do postaci, ze wzorem określającym funkcję gęstości radialnej z wyjątkiem wartości przedwykładniczego współczynnika liczbowego. Liczba 20000 znacznie przekracza oczekiwaną teoretycznie wielkość 4*pi. Ten wynik można zinterpretować, jako koszt obliczeniowy poniesiony w związku z zastosowaniem metody Monte Carlo i błądzenia wedle algorytmu Metropolisa, gdyż dokładność obliczenia energii całkowitej układu wzrasta wraz ze wzrostem liczby kroków błądzenia.
Na podstawie uzyskanych wyników proponujemy wykorzystać wzór 4 do obliczania energii układu. Oznacza to, że w obliczeniach energii całkowitej układów kwantowych z wykorzystaniem energii lokalnych oraz metody Monte Carlo i funkcji gęstości radialnej, stosowanie algorytmu Metropolisa (błądzenia pseudoprzypadkowego) nie jest konieczne. Dodatkowo, w przypadkach uzasadnionych kosztem obliczeń i symetrią układu, metodę Monte Carlo (losowanie punktów) można zastąpić systematycznym wyborem punktów, w których oblicza się energię lokalną a następnie całkowitą energię badanego układu. Zagadnienie to będzie przedmiotem następnej pracy.
W celu obliczenia tytułowej energii wykorzystamy uprzednio wyprowadzony wzór postaci 1 zawierający energię lokalną i funkcję gęstości radialnej:
 dla funkcji exp(-cr). Zastosowana metoda nie wykorzystuje algorytmu Metropolisa. Przedmiotem losowania (metoda Monte Carlo) będą wartości promienia wodzącego r elektronu w zakresie od zera do rmax. Sprawdzimy wyniki obliczeń dla zbioru wartości zmiennej wariacyjnej c i określimy jakość obliczenia energii dla różnych liczb przeprowadzonych losowań. Omawiane zagadnienia realizuje program MCnoMet.bas
dla funkcji exp(-cr). Zastosowana metoda nie wykorzystuje algorytmu Metropolisa. Przedmiotem losowania (metoda Monte Carlo) będą wartości promienia wodzącego r elektronu w zakresie od zera do rmax. Sprawdzimy wyniki obliczeń dla zbioru wartości zmiennej wariacyjnej c i określimy jakość obliczenia energii dla różnych liczb przeprowadzonych losowań. Omawiane zagadnienia realizuje program MCnoMet.bas

Tabela 1. przedstawia wartość liczbową energii minimalnej i wartości zmiennej wariacyjnej c w zależności od liczby losowań (llos).
W celu obliczenia tytułowej energii wykorzystamy uprzednio wyprowadzony wzór postaci 1 zawierający energię lokalną i funkcję gęstości radialnej:
 dla funkcji exp(-cr). Zastosowana tu metoda nie wykorzystuje algorytmu Metropolisa i rezygnuje metody Monte Carlo na korzyść systematycznego wzrostu wartości promienia wodzącego r elektronu w zakresie od rmin do rmax. Sprawdzimy wyniki obliczeń dla zbioru wartości zmiennej wariacyjnej c. Omawiane zagadnienia realizuje program noMCnoMe.bas:
dla funkcji exp(-cr). Zastosowana tu metoda nie wykorzystuje algorytmu Metropolisa i rezygnuje metody Monte Carlo na korzyść systematycznego wzrostu wartości promienia wodzącego r elektronu w zakresie od rmin do rmax. Sprawdzimy wyniki obliczeń dla zbioru wartości zmiennej wariacyjnej c. Omawiane zagadnienia realizuje program noMCnoMe.bas:
 W podsumowaniu należy stwierdzić, że dla trywialnego zagadnienia, jakim jest model atomu wodoru, zastosowanie wzoru 1 pozwala zmniejszyć znacznie koszt obliczeń w porównaniu do algorytmu Metropolisa. Otwartym pozostaje pytanie, czy ta korzyść występuje również w wypadku zagadnień nietrywialnych.
W podsumowaniu należy stwierdzić, że dla trywialnego zagadnienia, jakim jest model atomu wodoru, zastosowanie wzoru 1 pozwala zmniejszyć znacznie koszt obliczeń w porównaniu do algorytmu Metropolisa. Otwartym pozostaje pytanie, czy ta korzyść występuje również w wypadku zagadnień nietrywialnych.
'Program liczno.bas
DECLARE FUNCTION fufal! (x!, y!, z!)
DECLARE FUNCTION GRNF! () 'gaussowski generator pseudolosowy
'jednostki atomowe
CLS
llos = 500000 'liczba losowan
DIM SHARED c 'globalna zmienna wariacyjna
DIM x(800) 'wektor licznosci promieni
c = 1! 'wybrana wartosc zm. wariacyjnej
OPEN "dane.DAT" FOR APPEND AS #1
x = 0: y = 1.2: z = 0 'punkt startowy
FOR i = 1 TO llos
xzapas = x 'zabezpieczenie starych wspolrzednych
yzapas = y
zzapas = z
psi = fufal(x, y, z) 'wartosc psi w punkcie (x,y,z)
psikw = psi * psi 'kwadrat psi
'Losowanie nowego punktu (xn,yn,zn)
xn = 2 * RND - 1 'zakres (-1, 1)
yn = 2 * RND - 1
zn = 2 * RND - 1
pr = SQR(xn*xn + yn*yn + zn*zn) 'promien punktu (xn,yn,zn)
skok = .6 * GRNF 'skok przypadkowy wg rozkladu Gaussa
x = x + xn * skok / pr 'poprawione wspolrzedne nowego punktu
y = y + yn * skok / pr
z = z + zn * skok / pr
psinowa = fufal(x, y, z) 'wartsc psi w nowym punkcie
psinowakw = psinowa * psinowa
'obliczanie warunku przejscia lancucha do nowego punktu
rel = psinowakw / psikw 'stosunek nowej i starej psi
IF rel < 1 THEN 'przejscie bezwarunkowe gdy rel >= 1
IF RND > rel THEN 'przejscie odrzucone
x = xzapas 'przywrocenie starych wartosci
y = yzapas
z = zzapas
psinowa = fufal(x, y, z)
END IF
END IF
rn = SQR(x * x + y * y + z * z) 'promien do zliczania
liczba = INT(rn * 100) ' definicja adresu wektora promieni
x(liczba) = x(liczba) + 1 ' zliczanie promieni
NEXT i
FOR i = 1 TO 620
PRINT #1, i / 100, x(i)
NEXT i
CLOSE
END
FUNCTION fufal (x, y, z)
r = SQR(x * x + y * y + z * z)
fufal = EXP(-c * r)
END FUNCTION
FUNCTION GRNF
r1 = SQR(-LOG(1 - RND))
GRNF = r1 * SIN(2 * 3.14159265# * RND)
END FUNCTION

Na podstawie uzyskanych wyników proponujemy wykorzystać wzór 4 do obliczania energii układu. Oznacza to, że w obliczeniach energii całkowitej układów kwantowych z wykorzystaniem energii lokalnych oraz metody Monte Carlo i funkcji gęstości radialnej, stosowanie algorytmu Metropolisa (błądzenia pseudoprzypadkowego) nie jest konieczne. Dodatkowo, w przypadkach uzasadnionych kosztem obliczeń i symetrią układu, metodę Monte Carlo (losowanie punktów) można zastąpić systematycznym wyborem punktów, w których oblicza się energię lokalną a następnie całkowitą energię badanego układu. Zagadnienie to będzie przedmiotem następnej pracy.
W celu obliczenia tytułowej energii wykorzystamy uprzednio wyprowadzony wzór postaci 1 zawierający energię lokalną i funkcję gęstości radialnej:
'program liczy energię ale bez algorytmu Metropolisa
'jednostki atomowe
DECLARE FUNCTION fufal! (r!)
DIM SHARED c 'zmienna globalna
pi = 3.1415 'ludolfina
llos = 5000 'liczba losowan promienia
rmax = 5 'maksymalna dlugosc promienia
OPEN "energie.dat" FOR APPEND AS #1
CLS
SCREEN 12
FOR c = .1 TO 2 STEP .1 'obl. energii jako funkcja zm. war. c
Energia = 0
suma = 0
FOR i = 1 TO llos
r = RND * rmax
ro = fufal(r) * fufal(r) 'gestosc elektronowa
roradial = 4 * pi * r * r*ro 'funkcja gestosci radialnej
'Hpsi/psi*roradial
Elokalna = (-c * c / 2 + (c - 1) / r) * roradial
Energia = Energia + Elokalna
suma = suma + roradial
NEXT i
PRINT #1, c, Energia / suma
NEXT c
CLOSE
FUNCTION fufal (r)
fufal = EXP(-c * r)
END FUNCTION
Rys. 1 przedstawia wyniki działania programu dla 5000 losowań
Tabela 1. przedstawia wartość liczbową energii minimalnej i wartości zmiennej wariacyjnej c w zależności od liczby losowań (llos).
llos c Energia
10 1.1 -0.5174
100 1.0 -0.5000001
500 1.0 -0.4999999
1000 1.0 -0.5000001
2000 1.0 -0.5000001
5000 1.0 -0.5000000
7000 1.0 -0.4999999
Wyniki pokazują, że już sto losowań wartości promienia daje dokładną wartość energii (do obliczeń użyliśmy zmiennych pojedynczej precyzji) dla prawidłowej wartości zmiennej wariacyjnej c = 1.W celu obliczenia tytułowej energii wykorzystamy uprzednio wyprowadzony wzór postaci 1 zawierający energię lokalną i funkcję gęstości radialnej:
'Author Wojciech Szczepankiewicz
'Silesian University of Technology
'Gliwice, Poland
DECLARE FUNCTION fufal! (r!)
'atomic units
DIM SHARED c 'variational variable
pi = 3.1415 'ludolphine
rmin = .01 'min radius
rmax = 5 'max radius
OPEN "data.dat" FOR APPEND AS #1
CLS
'energy calc. as a function of variational variable
FOR c = .1 TO 2 STEP .05
Energy = 0
sum = 0
FOR r = rmin TO rmax STEP .01
rhoradial = fufal(r) * fufal(r) 'density
freq = 4 * pi * r * r * rhoradial 'radial density function
'Hpsi/psi*rhoradial
Elocal = (-c * c / 2 + (c - 1) / r) * freq
Energy = Energy + Elocal
sum = sum + freq
NEXT r
PRINT #1, c, Energy / sum
NEXT c
CLOSE
FUNCTION fufal (r)
fufal = EXP(-c * r)
END FUNCTION
Rys. 1 przedstawia wyniki działania programu dla kolejnych 499 punktów promienia
Brak komentarzy :
Prześlij komentarz